Interesting question lately on a common syndrome. Many sources neither perform MRI not treat Rolandic Epilepsy. This is appropriate and data based.But is it Rolandic? There are
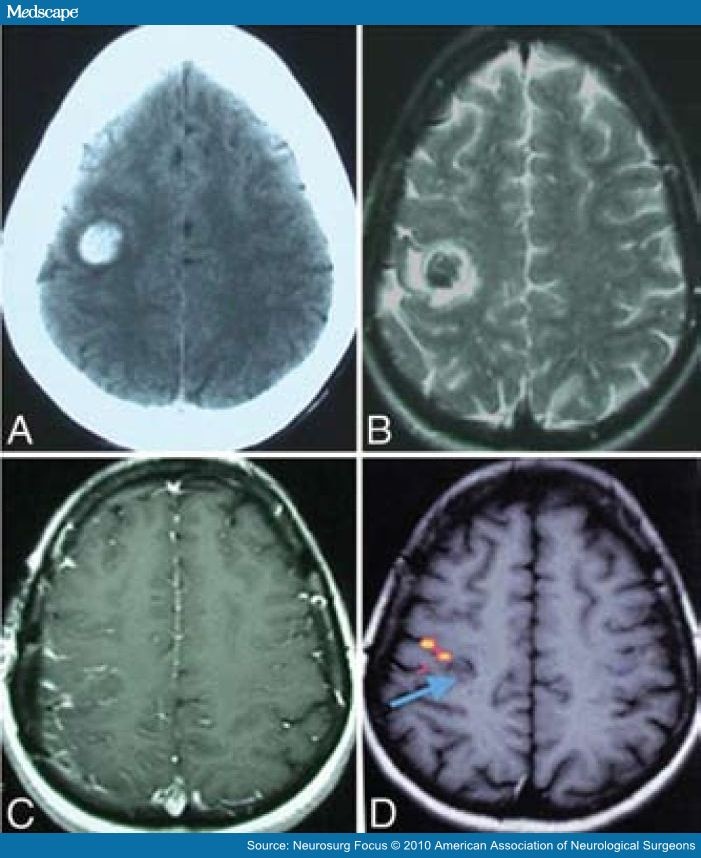 atypical features based on history, physical and EEG.
atypical features based on history, physical and EEG.
Other considerations include...
- Rolandic spikes and no seizures (often with behavior problems, headaches, or autonomic dysfunction)
- Rolandic spikes and a history of antecedent brain damage, cerebral palsy, or active local pathology
- Central spikes occurring commonly in Rett syndrome and fragile X syndrome
- Malignant rolandic epilepsy
- Psychomotor seizures and evolving temporal lobe epilepsy
- The aphasia-convulsion (Landau-Kleffner) syndrome and massive mid temporal spikesJR
Benign Epilepsy of Childhood With Centrotemporal Spikes
A benign partial epilepsy of childhood, this condition, benign epilepsy of childhood with centrotemporal spikes (BECCT), is defined within the International League Against Epilepsy (ILAE) classification scheme as an idiopathic age- and localization-related epileptic syndrome with a combination of clinical and EEG characteristics used for diagnosis.
This epileptic syndrome is characterized by brief, simple partial and hemifacial motor seizures with associated somatosensory symptoms, which have a tendency to evolve into generalized tonic-clonic seizures. EEG shows high-voltage centrotemporal spikes often followed by a slow wave. BECCT is also known as lingual epilepsy, sylvian seizures, benign centrotemporal epilepsy, and benign rolandic epilepsy.
If the patient has the typical clinical history and EEG findings and has normal findings on neurologic examination, further workup is not indicated. However, in the presence of atypical features or abnormal examination findings, the use of magnetic resonance imaging (MRI) is indicated.[29]
Benign rolandic epilepsy has been reported to occur in the presence of CNS pathology. However, in most of these reported instances, the BECCT was probably coincidental.
Epidemiology
BECCT is the most common epilepsy syndrome in childhood. In Connecticut, USA, it represents 9.6% of all epilepsies in children aged 0-15 years. However, BECCT and its variants may represent 20-25% of epilepsy cases diagnosed in patients aged 5-15 years.
Studies from other countries show that BECCT accounts for 6.5-16% of all childhood epilepsy. In a study from India, however, it represented only 1.6% of epilepsies in children younger than age 16 years. In a study from Italy, epilepsies with rolandic foci accounted for up to 23.9% of epilepsies in children aged 4-15 years.
Reported incidence of seizures with central temporal spikes ranges from 10.7-21 per 100,000. Age of onset ranges from 2-13 years but usually is between 4 and 11 years, and frequency of onset peaks at 5-9 years. The disorder occurs more commonly in boys; the boy-to-girl ratio is 6:4. A study by Kramer et al found no gender difference in incidence.[30]...
Contributor Information and Disclosures
Author
Ahmad K Kaddurah, MD Assistant Professor, Department of Pediatrics and Human Development, Michigan State University College of Human Medicine; Director of Pediatric Neurology, Hurley Medical Center
Ahmad K Kaddurah, MD is a member of the following medical societies: American Academy of Neurology, American Academy of Pediatrics, American Clinical Neurophysiology Society, American Medical Association, and Child Neurology Society
Disclosure: Nothing to disclose.
Ahmad K Kaddurah, MD is a member of the following medical societies: American Academy of Neurology, American Academy of Pediatrics, American Clinical Neurophysiology Society, American Medical Association, and Child Neurology Society
Disclosure: Nothing to disclose.
Coauthor(s)
Bhagwan I Moorjani, MD, FAAP, FAAN Consulting Staff, Department of Neuroscience, Director, Department of Neuroscience, Division of Evoked Response Laboratory, Children's National Medical Center
Bhagwan I Moorjani, MD, FAAP, FAAN is a member of the following medical societies: American Academy of Neurology
Disclosure: Nothing to disclose.
Bhagwan I Moorjani, MD, FAAP, FAAN is a member of the following medical societies: American Academy of Neurology
Disclosure: Nothing to disclose.
Specialty Editor Board
James J Riviello Jr, MD George Peterkin Endowed Chair in Pediatrics, Professor of Pediatrics, Section of Neurology and Developmental Neuroscience
No comments:
Post a Comment